Sex chromosome evolution and hybridization between lineages having different sex-determining regions
Work in progress. Postdoc research in the Merilä lab
Sex chromosomes are highly conserved in mammals and birds but remain labile in other vertebrates such as amphibians and fishes where different sex chromosomes can be found between closely related lineages. The high diversity of sex chromosomes in these taxa result from frequent sex chromosome turnovers, yet little is known about the underlying evolutionary mechanisms and many species still have unknown sex chromosomes. Here we use nine-spined sticklebacks (Pungitius pungitius) as a model system to study the mechanisms of labile sex chromosome evolution and turnover. Using whole-genome resequencing data, I identified the previously unknown homomorphic sex chromosomes in the western European lineage (WL) and indicate a putative inversion on the Y chromosome. In addition, I showed that this homomorphic sex chromosome takes over the heteromorphic sex chromosome (LG12) in the hybrid zone between western (WL) and eastern (EL) lineages, indicating an ongoing sex chromosome turnover potentially driven by selection.
Work in progress. Postdoc research in the Merilä lab
Sex chromosomes are highly conserved in mammals and birds but remain labile in other vertebrates such as amphibians and fishes where different sex chromosomes can be found between closely related lineages. The high diversity of sex chromosomes in these taxa result from frequent sex chromosome turnovers, yet little is known about the underlying evolutionary mechanisms and many species still have unknown sex chromosomes. Here we use nine-spined sticklebacks (Pungitius pungitius) as a model system to study the mechanisms of labile sex chromosome evolution and turnover. Using whole-genome resequencing data, I identified the previously unknown homomorphic sex chromosomes in the western European lineage (WL) and indicate a putative inversion on the Y chromosome. In addition, I showed that this homomorphic sex chromosome takes over the heteromorphic sex chromosome (LG12) in the hybrid zone between western (WL) and eastern (EL) lineages, indicating an ongoing sex chromosome turnover potentially driven by selection.
UCE-derived mitochondrial phylogeny of serotine bats (genus Eptesicus)
Work in progress. In collaboration with Dr. Anderson Feijó and Dr. Burton K. Lim
Studies of evolution and biodiversity require a good understanding of species relationships using multilocus molecular phylogenies. Studies of many non-model organisms have been limited to a few nuclear genes or mitochondrial genes that always show discordant phylogenies indicating complex evolutionary histories. Here we use the program MitoFinder to identify and annotate mitochondrial sequences from published UCE libraries of bats, and construct the hitherto most taxa-complete mitochondrial phylogeny of Eptesicus (Histiotus) bats by combining annotated sequences, NCBI references, and a few newly sequenced individuals from previously understudied areas. We showed pervasive mito-nuclear discordances in the genus Eptesicus, indicating complex evolutionary histories that include repeated introgression across lineages. In addition, we updated the mitochondrial phylogeny and systematics of the clade Eptesicus (Histiotus) and indicated still underestimated diversity of these fascinating species.
Work in progress. In collaboration with Dr. Anderson Feijó and Dr. Burton K. Lim
Studies of evolution and biodiversity require a good understanding of species relationships using multilocus molecular phylogenies. Studies of many non-model organisms have been limited to a few nuclear genes or mitochondrial genes that always show discordant phylogenies indicating complex evolutionary histories. Here we use the program MitoFinder to identify and annotate mitochondrial sequences from published UCE libraries of bats, and construct the hitherto most taxa-complete mitochondrial phylogeny of Eptesicus (Histiotus) bats by combining annotated sequences, NCBI references, and a few newly sequenced individuals from previously understudied areas. We showed pervasive mito-nuclear discordances in the genus Eptesicus, indicating complex evolutionary histories that include repeated introgression across lineages. In addition, we updated the mitochondrial phylogeny and systematics of the clade Eptesicus (Histiotus) and indicated still underestimated diversity of these fascinating species.
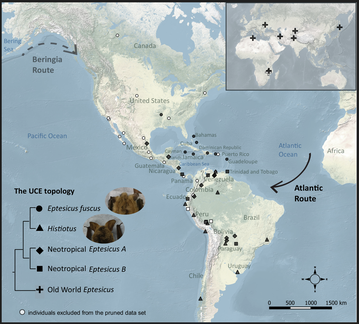
Phylogenetics of the bat genus Eptesicus using UCEs
Ph.D. project published in: https://doi.org/10.1016/j.ympev.2022.107582
The bat genus Eptesicus is distributed globally and yet the phylogenetic relationships among species within this genus, especially in the New World, is not clear. Interestingly, molecular studies found that the New World Eptesicus are more closely related to Histiotus, another morphological genus having enlarged ears, than to the Old World Eptesicus. To better estimate the phylogeny of the New World Eptesicus and Histiotus species, we collected samples across taxonomic and geographic scales and genomic markers of thousands of ultra-conserved elements (UCEs). We further reconstructed ancestral distribution and tested hypotheses about the initial dispersal route (on-land versus trans-marine) of Eptesicus bats to colonize the New World. I sincerely thank all the museums and researchers who kindly provided samples to support this study!
Ph.D. project published in: https://doi.org/10.1016/j.ympev.2022.107582
The bat genus Eptesicus is distributed globally and yet the phylogenetic relationships among species within this genus, especially in the New World, is not clear. Interestingly, molecular studies found that the New World Eptesicus are more closely related to Histiotus, another morphological genus having enlarged ears, than to the Old World Eptesicus. To better estimate the phylogeny of the New World Eptesicus and Histiotus species, we collected samples across taxonomic and geographic scales and genomic markers of thousands of ultra-conserved elements (UCEs). We further reconstructed ancestral distribution and tested hypotheses about the initial dispersal route (on-land versus trans-marine) of Eptesicus bats to colonize the New World. I sincerely thank all the museums and researchers who kindly provided samples to support this study!

My visit to the Smithsonian National Museum of Natural History. |
|
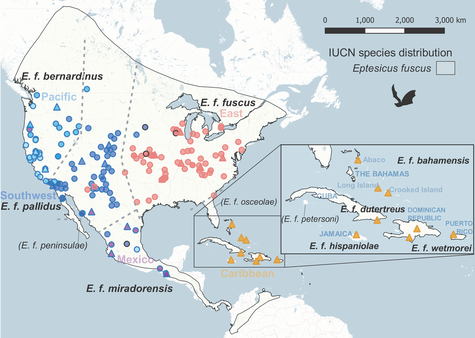
Range-wide phylogeography of the big brown bat (Eptesicus fuscus) using RADseq
Ph.D. project published in: https://doi.org/10.1111/jbi.14362
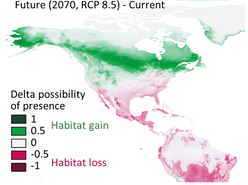
The big brown bat (Eptesicus fuscus) is a common house bat in North America, distributed from southern Canada to norther South America including most of the Caribbean Islands. The previous study showed mitochondrial divergence but a lack of nuclear structure across the distribution range. To test whether gene flow has eroded the historical divergence (such as due to glacial isolation) in the nuclear genome, we collected range-wide samples and genome wide markers using the restriction site-associated DNA sequencing (RADseq). Analyses of population genetics, phylogeography, and demography supported historical isolation followed by secondary gene flow, showing signals of fine-scale nuclear divergence despite effects of on-going gene flow. In addition, species distribution modeling predicted further northward range shifts of this species under the future climate change.
Ph.D. project published in: https://doi.org/10.1111/jbi.14362
The big brown bat (Eptesicus fuscus) is a common house bat in North America, distributed from southern Canada to norther South America including most of the Caribbean Islands. The previous study showed mitochondrial divergence but a lack of nuclear structure across the distribution range. To test whether gene flow has eroded the historical divergence (such as due to glacial isolation) in the nuclear genome, we collected range-wide samples and genome wide markers using the restriction site-associated DNA sequencing (RADseq). Analyses of population genetics, phylogeography, and demography supported historical isolation followed by secondary gene flow, showing signals of fine-scale nuclear divergence despite effects of on-going gene flow. In addition, species distribution modeling predicted further northward range shifts of this species under the future climate change.
|

|
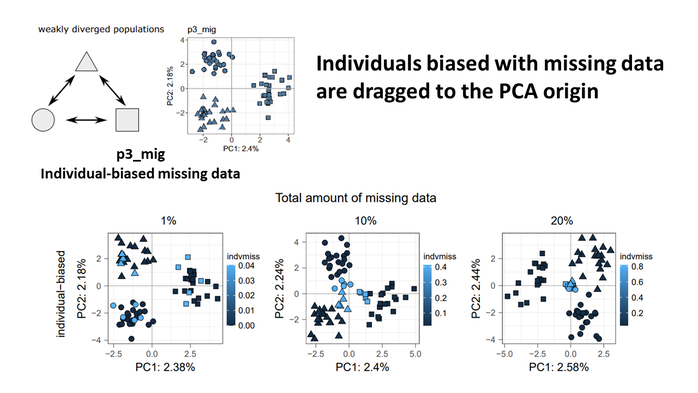
Nonrandom missing data can bias Principal Component Analysis inference of population genetic structure
Ph.D. project published in: https://onlinelibrary.wiley.com/doi/abs/10.1111/1755-0998.13498
Principal Component Analyses (PCA) have been widely used to illustrate population structure and the overall diversification pattern in studies of population genetics, such as using next-generation sequencing (NGS). However, NGS methods tend to generate lots of missing data that could affect downstream analyses and bias the interpretation of PCA plots. Here we showed that when missing data are nonrandomly distributed across individuals (or populations), the individuals having more missing data tend to be dragged away from their true population cluster and towards the origin of the PCA plot generated using the default mean-imputation. We suggest that researchers always double-check the potential missing data bias by plotting PCA results with a color gradient showing per sample missingness, and interpret samples close to PCA origin with extra caution as they can be missing data biased or admixed. In addition, we showed that it is important to explore different filtering parameters and use complementary analyses to infer population structure, especially in non-model organisms that tend to have varying sample quality and unclear genetic divergence. Codes used can be found on Github (https://github.com/xuelingyi/missing_data_PCA).
Ph.D. project published in: https://onlinelibrary.wiley.com/doi/abs/10.1111/1755-0998.13498
Principal Component Analyses (PCA) have been widely used to illustrate population structure and the overall diversification pattern in studies of population genetics, such as using next-generation sequencing (NGS). However, NGS methods tend to generate lots of missing data that could affect downstream analyses and bias the interpretation of PCA plots. Here we showed that when missing data are nonrandomly distributed across individuals (or populations), the individuals having more missing data tend to be dragged away from their true population cluster and towards the origin of the PCA plot generated using the default mean-imputation. We suggest that researchers always double-check the potential missing data bias by plotting PCA results with a color gradient showing per sample missingness, and interpret samples close to PCA origin with extra caution as they can be missing data biased or admixed. In addition, we showed that it is important to explore different filtering parameters and use complementary analyses to infer population structure, especially in non-model organisms that tend to have varying sample quality and unclear genetic divergence. Codes used can be found on Github (https://github.com/xuelingyi/missing_data_PCA).